
In late-December 2019, the coronavirus disease 2019 (COVID-19) was first detected in Wuhan City, China, causing an easily transmissible respiratory illness. By March 2020, the World Health Organization (WHO) declared its outbreak a global pandemic.
Caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), COVID-19 has infected over 107.48 million people globally. To date, more than 2.35 million have already died.
As the pandemic has evolved, the virus has mutated into variants that are more easily transmissible and may potentially cause more severe symptoms.
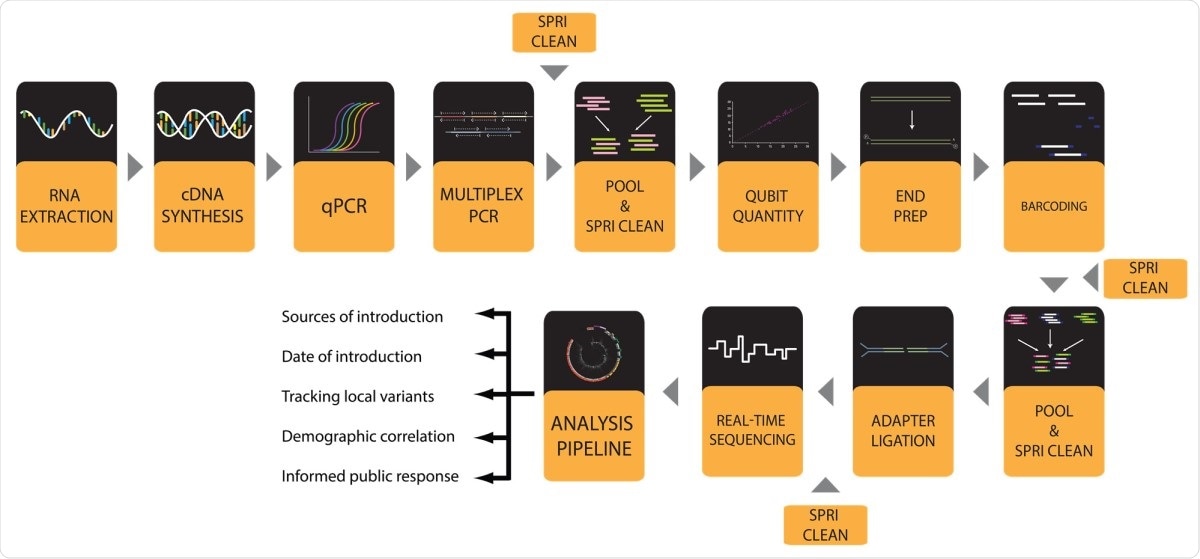
Now, researchers at the Southern Illinois University, U.S.A., presented a comprehensive and well-validated set of working protocols for sequencing high-quality SARS-CoV-2 genomes in high throughput from patient samples. This can help in conducting viral genome sequencing, which can help determine mutations of the virus and help governments plan in implementing measures to stem the pandemic.
Viral genome sequencing
It is crucial to perform viral genome sequencing to help address any potential problems that may arise with new mutations of the virus. Viruses can evolve quickly, which can result in heightened transmissibility or virulence and pose a serious threat to people worldwide.
The SARS-CoV-2 has two new variants that are more transmissible and may potentially cause more severe symptoms. The U.K. variant has already spread to other countries, causing skyrocketing cases in places like the U.S., while the South African variant may evade the antibodies produced due to vaccination.
Hence, viral genome sequencing can delineate conserved and mutable regions that could provide valuable insights into the development of effective vaccines and treatments for SARS-CoV-2.
Currently, there are many various approaches to sequencing SARS-CoV-2 genomes, such as metagenomic sequencing, PCR-amplification based methods, and targeted enrichment sequencing. Two of these methods, particularly the metagenomic and target genomes, are expensive to generate large-scale complete genomes.
Meanwhile, a PCR-based amplification may be a potential solution to address the limitations of the other methods. The PCR-based amplification method amplifies the target of interest at the same time.
The study
In the current study, published on the bioRxiv* preprint server, the researchers introduced a method to sequence high-quality SARS-CoV-2 genomes in a high throughout from patient samples. The research team’s laboratory adopted and modified the protocols, which were developed by the ARTIC Network to sequences SARS-CoV-2 viral genomes utilizing the MiniION nanopore sequencing platform from Oxford Nanopore Technologies (ONT).
From there, the team presented an entire validated sequence, including a how-to guide for every step from viral ribonucleic acid (RNA) extraction to data visualization. The team validated the new protocols through the successful processing of more than 1,000 samples and sequencing more than 600 high-quality SARS-CoV-2 genomes from Illinois.
To arrive at the study findings, the researchers performed the entire workflow to produce SARS-CoV-2 genomes in a 96-well plate format. It starts with extracting RNA from viral transport media (VTM) that contains patient-derived nasopharyngeal (NP) swabs. Then, the team converts the extracted RNA to complementary DNA (cDNA), which is utilized for the approximation of viral load by quantitative PCR (qPCR).
After conducting qPCR, the researchers amplified viral genome cDNA in 96-well format to reduce contaminating the sequences and to boost the copy number of viral genomes. In this method, the researchers used two pools of primers designed by the ARTIC Network.
After, the team performed a multiplex PCR with two pools of ARTIC primers, and the two separate PCR product pools for each sample are pooled, while the amplicons are isolated via a clean-up step.
Lastly, genomes with adequate coverage can be seen and observed by phylogenetic analyses and variant calling done by using web-based tools like Nextstrain.
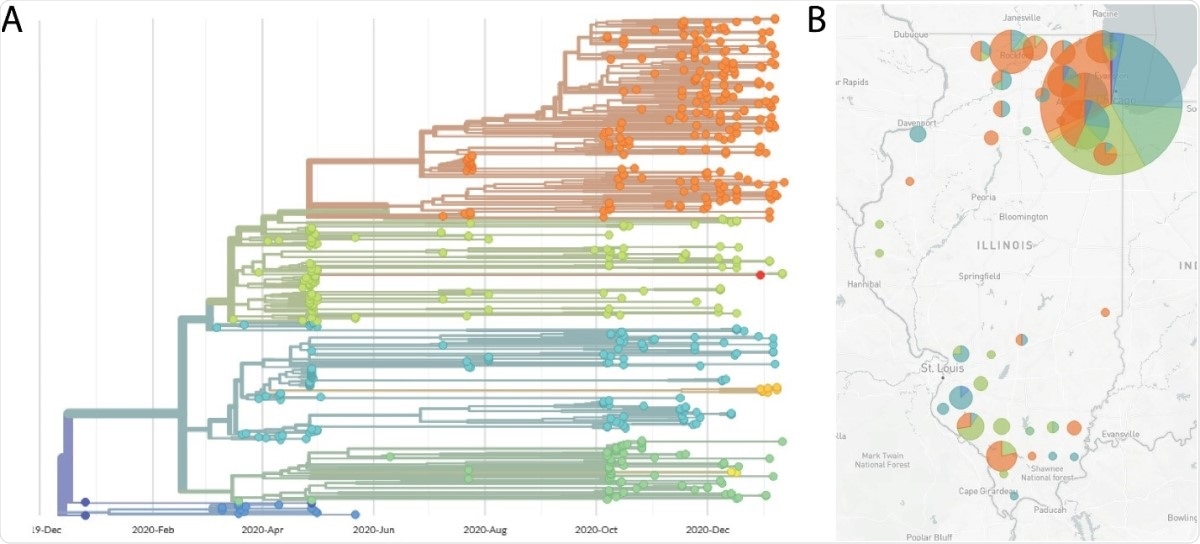
Hence, the protocol can provide an easier way to perform sequencing. The protocols implement the ARTIC PCR and MinION nanopore to sequence 96 samples at a time.
The team believes that the new protocol can provide a reliable starting point for those who want to produce SARS-CoV-2 genome sequences using nanopore sequencing technology.
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU) – https://gisanddata.maps.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6
- Pater, A., Bosmeny, M., Parasrampuria, M. et al. (2021). High Throughput Nanopore Sequencing of SARS-CoV-2 Viral Genomes from Patient Samples. bioRxiv. doi: https://doi.org/10.1101/2021.02.09.430478, https://www.biorxiv.org/content/10.1101/2021.02.09.430478v1
Posted in: Medical Science News | Medical Research News | Disease/Infection News
Tags: Antibodies, Coronavirus, Coronavirus Disease COVID-19, DNA, Genome, Genomic, Genomic Sequencing, High Throughput, Laboratory, Pandemic, Phylogeny, Research, Respiratory, Respiratory Illness, Ribonucleic Acid, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome, Virus

Written by
Angela Betsaida B. Laguipo
Angela is a nurse by profession and a writer by heart. She graduated with honors (Cum Laude) for her Bachelor of Nursing degree at the University of Baguio, Philippines. She is currently completing her Master's Degree where she specialized in Maternal and Child Nursing and worked as a clinical instructor and educator in the School of Nursing at the University of Baguio.
Source: Read Full Article

