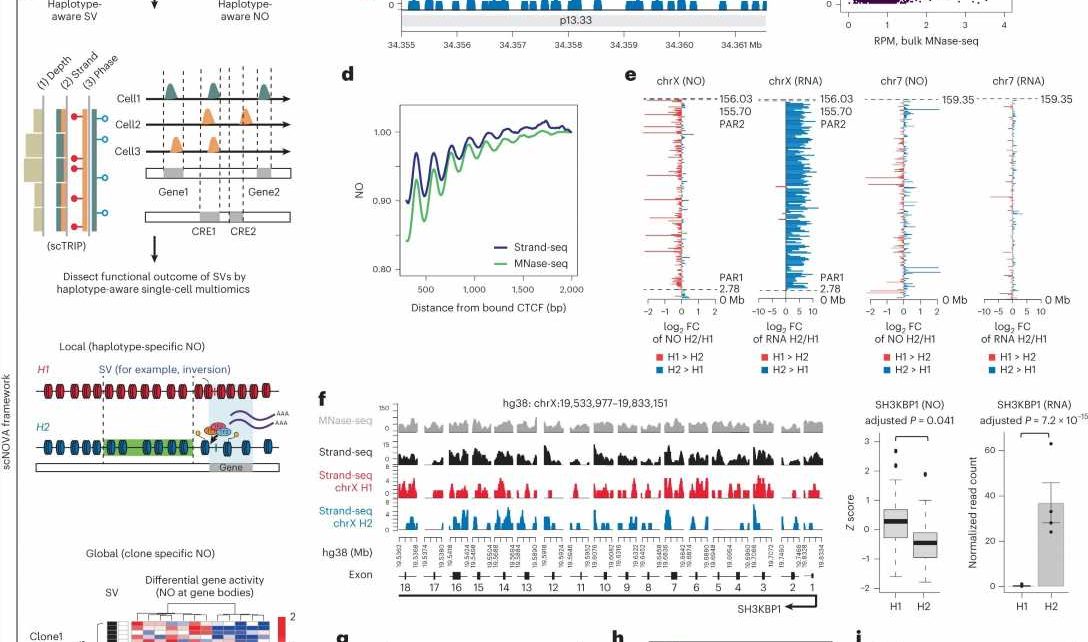
 and globally (clone-specific NO). Orange, Strand-seq reads mapped to the Watson (W) strand; green, reads mapped to the Crick (C) strand. b , Strand-seq-based NO tracks in NA12878 reveal nucleosome positions well-concordant with bulk MNase-seq, depicted for a chromosome 12 locus with relatively regular nucleosome positioning. Red, NO tracks mapping to haplotype 1 (H1); blue, H2; black, combining phased and unphased reads; gray, MNase-seq. The y axis depicts the mean read counts at each bp in 10 bp bins. c , Correlated NO at consensus DNase I hypersensitive sites for NA12878. d , Averaged nucleosome patterns at CTCF binding sites in NA12878, using pseudobulk Strand-seq and MNase-seq. e , FCs of haplotype-resolved NO in gene bodies plotted for chromosome X and chromosome 7 (a representative autosome) in NA12878. FCs of haplotype-resolved RNA expression measurements are shown to the right. f , Pseudobulk haplotype-phased NO track of exons of the representative chromosome X gene SH3KBP1 based on Strand-seq. Boxplots comparing H1 and H2 use two-sided Wilcoxon rank sum tests followed by Benjamini–Hochberg multiple testing (FDR) correction (boxplots defined by minima = 25th percentile – 1.5 × interquartile range (IQR), maxima = 75th percentile + 1.5 × IQR, center = median and bounds of box = 25th and 75th percentile; n = 47 single cells). Bar charts show haplotype-specific RNA expression of SH3KBP1 (two-sided likelihood ratio test followed by FDR correction; n = 4 biological replicates; data are presented as mean values ± s.e.m.). g , Inverse correlation of NO at gene bodies and gene expression. NO is based on pseudobulk Strand-seq libraries from RPE-1. Gene bodies were scaled to the same length. h , Cell-typing based on NO at gene bodies (AUC = 0.96). Cell line codes: Blue, RPE-1; Purple, BM510; Magenta, C7; LV, latent variable. i , Receiver operating characteristics for inferring altered gene activity by analyzing NO at gene bodies, using pseudobulk Strand-seq libraries from in silico cell mixing. Credit: Nature Biotechnology (2022). DOI: 10.1038/s41587-022-01551-4")
Cancer has many faces—no wonder, then, that the range of cancer-causing mutations is huge as well. The totality of such genomic alterations in an individual is what experts call a “mutational landscape.” These landscapes differ from one another depending on the type of cancer. And even people suffering from the same cancer often have different mutation patterns.
Researchers have already cataloged the mutational landscapes of numerous types of cancer. Somatic structural variants (SVs) have been shown to account for more than half of all cancer-driving mutations. These are those mutations in cells that emerge over the course of life—such as when copying errors creep into the DNA during cell division—and thereby alter the chromosome structure.
They are not inherited and are found only in affected cells and in their daughter cells. As we age, such genomic alterations become more numerous, and a person’s mutational landscape increasingly comes to resemble a unique mosaic.
Although somatic SVs play a crucial role in cancer development, relatively little is known about them. “There is a lack of methods that analyze their effects on cell function,” explains Dr. Ashley Sanders, who heads the Genome Stability and Somatic Mosaicism Lab at the Max Delbrück Center. That’s changing thanks to new research findings, which Sanders recently published in the journal Nature Biotechnology along with the European Molecular Biology Laboratory (EMBL).
“We developed a computational analysis method to detect and identify the functional effects of somatic SVs,” she reports. This enabled the team to understand the molecular consequences of individual somatic mutations in different leukemia patients, giving them new insights into the mutation-specific alterations. Sanders says it may also be possible to use these findings to develop therapies that target the mutated cells, adding that “they open up exciting new avenues for personalized medicine.”
Even more detailed than conventional single-cell analyses
Their calculations are based on data from Strand-seq—a special single-cell sequencing method that Sanders played an instrumental role in developing and that was first introduced to the scientific community in 2012. This technique can examine a cell’s genome in much greater detail than conventional single-cell sequencing technologies.
Thanks to a sophisticated experimental protocol, the Strand-seq method can independently analyze the two parental DNA strands (one from the father and one from the mother). With conventional sequencing methods, distinguishing such homologs—chromosomes that are similar in shape and structure but not identical—is nearly impossible.
“By resolving the individual homologs within a cell, somatic SVs can be identified much better than with other methods,” explains Sanders. The approach used for doing this was described by the researcher and her colleagues in a paper that appeared in Nature Biotechnology in 2020.
The research team is part of the joint research focus “Single-Cell Approaches for Personalized Medicine” of the Berlin Institute of Health at Charité (BIH), Charité—Universitätsmedizin Berlin, and the Max Delbrück Center.
Building on this work, they are now able to also determine the positions of nucleosomes in each cell. Nucleosomes are units of DNA wrapped around protein complexes called histones, and play a crucial role in organizing chromosomes. The position of nucleosomes can change during gene expression, with the type of wrapping revealing whether or not a gene is active. Sanders and her colleagues developed a self-learning algorithm to compare the gene activity of patient cells with and without somatic SV mutations, allowing them to determine the molecular impact of the structural variants.
New targets for cancer therapy
“We can now take a sample from a patient, look for the mutations that led to the disease, and also learn the signaling pathways that the disease-causing mutations disrupt,” explains Sanders. For example, the team was able to identify a rare but very aggressive mutation in a leukemia patient. The nucleosome analysis provided the researchers with information about the signaling pathways involved, which they used to specifically inhibit the growth of cells containing the mutation.
“This means that a single test tells us something about the cellular mechanisms involved in cancer formation,” says Sanders. “We can eventually use this knowledge to develop personalized treatments, guided by each patient’s unique condition.”
More information:
Hyobin Jeong et al, Functional analysis of structural variants in single cells using Strand-seq, Nature Biotechnology (2022). DOI: 10.1038/s41587-022-01551-4
Ester Falconer et al, DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution, Nature Methods (2012). DOI: 10.1038/nmeth.2206
Ashley D. Sanders et al, Single-cell analysis of structural variations and complex rearrangements with tri-channel processing, Nature Biotechnology (2019). DOI: 10.1038/s41587-019-0366-x
Journal information:
Nature Biotechnology
,
Nature Methods
Source: Read Full Article