
In a recent report posted to the bioRxiv* preprint server, researchers from the United Kingdom and Uganda reveal unique protein features of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) relative to other representatives of Sarbecoviruses.
The causative agent of the coronavirus disease 2019 (COVID-19), SARS-CoV-2, belongs to the Betacoronavirus genus and Sarbecovirus subgenus of the Coronaviridae family, which is a group of enveloped, single-stranded RNA viruses.
Other representatives of Sarbecoviruses include SARS-CoV (which was the coronavirus that led to the SARS outbreak in 2002 and 2003), but also a myriad of SARS-like bat coronaviruses that have been identified and analyzed in depth.
Among the viral structural proteins, the spike glycoprotein has a pivotal role in the host range, cell tropism, and entrance, as well as infectivity traits. Hence, it is no wonder that it is considered a prime target of the host immune response.
Therefore, a thorough comparative analysis of viral proteins would help immensely in our better understanding of viral biology and pathology, providing insights into its origin, as well as the conditions that led to the ongoing COVID-19 pandemic.
This is why Dr. Matthew Cotten, Dr. David L. Roberston, and Dr. My V.T. Phan from Uganda and the United Kingdom aimed to identify unique peptide regions of SARS-CoV-2 when compared to all available Sarbecoviruses in order to appraise the features that might enable SARS-CoV-2 to replicate and transmit efficiently among us.
.jpg)
Comparing genomes and appraising evolutionary distances
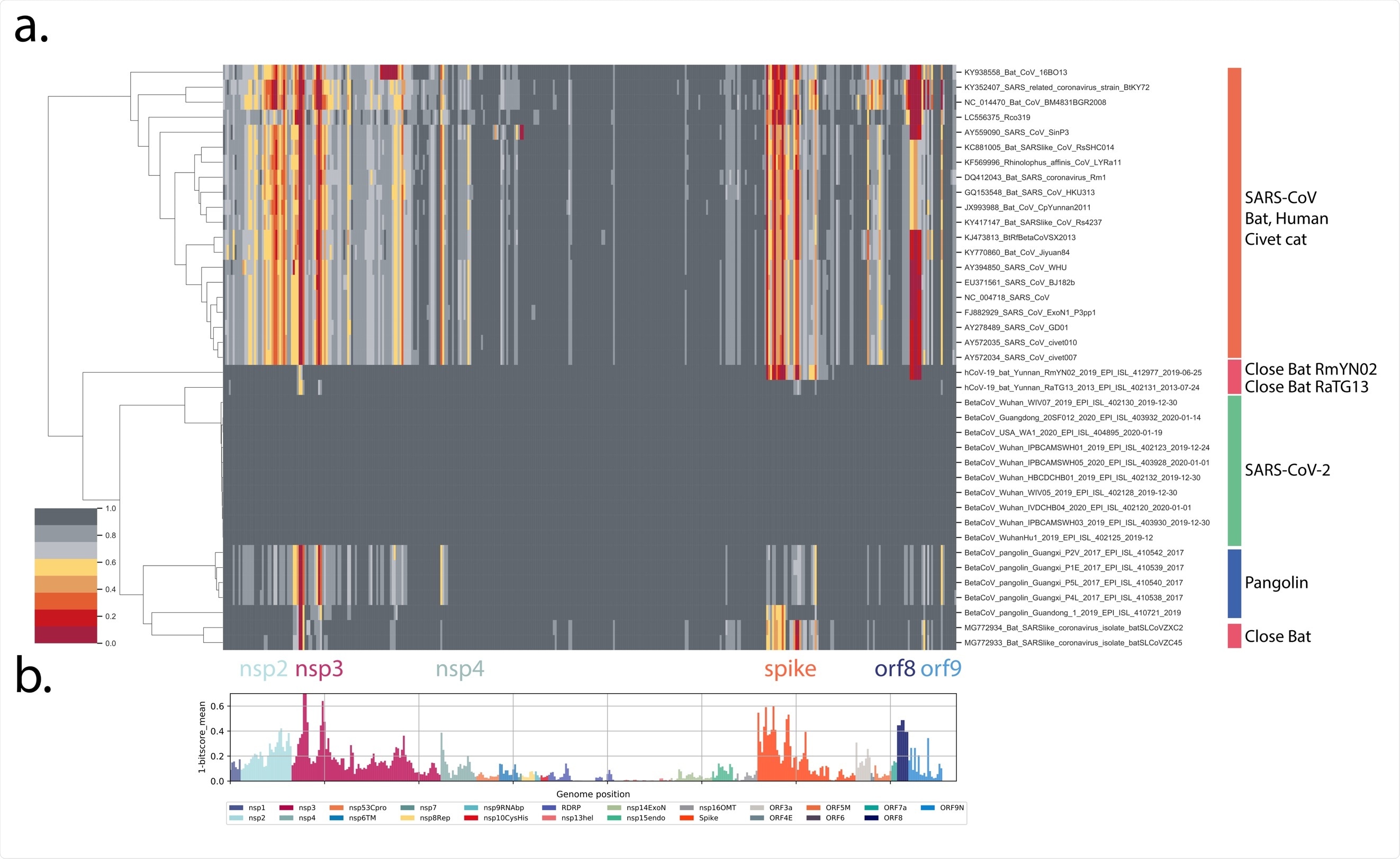
In a nutshell, this research group has explored the genomes across the Sarbecovirus subgenus by using profile hidden Markov models. This modeling approach is based on a statistical description of the properties of viral proteins and their amino acid sequences.
More specifically, ten early SARS-CoV-2 genomes were compared to a representative subset of Sarbecovirus genomes obtained from human individuals, bats, pangolins, and civet cats. These were selected after the same analysis has been applied to all available Betacoronavirus genomes in order to avoid missing any surprisingly close viral genome regions.
In order to appraise total domain distances between viral groups, normalized bit-score sums (grouped into SARS-CoV-2 and Sarbecoviruses from human, bat, pangolin and civet cat) were summed for all domains and for each genome.
Unique nature of SARS-CoV-2
Detected changes in spike glycoprotein of SARS-CoV-2 – in comparison to a large set of known Sarbecovirus – reveal that the recent zoonotic source of this virus is yet to be found but also underpin a rather unique nature of the SARS-CoV-2 genome.
In line with previous reports, a small set of bat and pangolin-derived Sarbecoviruses demonstrate the most significant similarity to SARS-CoV-2, while a measure of proteome similarity showed that it is unlikely that bat Sarbecoviruses are the direct source of the pandemic virus.
Furthermore, the regions of variance that were identified in this study may indicate either functional changes in SARS-CoV-2 proteins or amino acid positions that can be modified without compromising the requisite functions of the protein.
The detailed spike analysis revealed 82 domains of 15 amino acids that have demonstrated high variation in the Sarbecoviruses, while 29 of these domains show changes in variants of concern relative to the early lineage of SARS-CoV-2.
Being wary of the viral adaptation
This study gives credence to the notion of continuous genomic variant surveillance, which should then translate to the preparedness of vaccine producers to accommodate such spike glycoprotein changes in the next generation of vaccine updates.
"In broad terms, the SARS-CoV-2 evolution observed in the current variants of concern has sampled only 36% of the possible spikes changes which have occurred historically in Sarbecovirus evolution", say study authors in this bioRxiv paper.
"It is highly likely that a large number of new SARS-CoV-2 variants with changes in these regions are possible, compatible with virus replication and expected in the coming months unless global viral replication is severely reduced", they add.
In conclusion, such a high mutation rate of SARS-CoV-2 in combination with the remarkable number of SARS-CoV-2 infections worldwide gives rise to massive viral adaptation. Hence, further experiments will be necessary to discern true functional changes from neutral evolution.
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Cotten, M., Robertson, D.L. & Phan, M.V.T. Unique protein features of SARS-CoV-2 relative to other Sarbecoviruses. bioRxiv. https://doi.org/10.1101/2021.04.06.438675, https://www.biorxiv.org/content/10.1101/2021.04.06.438675v1
Posted in: Medical Research News | Disease/Infection News
Tags: Amino Acid, Cell, Coronavirus, Coronavirus Disease COVID-19, Evolution, Genome, Genomic, Glycoprotein, Immune Response, Mutation, Next Generation, Pandemic, Pathology, Peptides, Protein, Proteome, Research, Respiratory, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome, Vaccine, Virus

Written by
Dr. Tomislav Meštrović
Dr. Tomislav Meštrović is a medical doctor (MD) with a Ph.D. in biomedical and health sciences, specialist in the field of clinical microbiology, and an Assistant Professor at Croatia's youngest university – University North. In addition to his interest in clinical, research and lecturing activities, his immense passion for medical writing and scientific communication goes back to his student days. He enjoys contributing back to the community. In his spare time, Tomislav is a movie buff and an avid traveler.
Source: Read Full Article

